
Mapping Data
The mammoth data that deluges the bioinformatics domain could be majorly attributed to the next-generation sequencing experiments. Often times, it is required to elucidate the genomic source of the sequencing data. Although the species is known, we might still be interested to know which part of the genome is being represented by the sequenced reads. That will possibly enlighten us on say, the genes that are expressed in a particular phenotype. Broadly, mapping the reads to the reference genome is the process of data mapping.
Hypothetically, we need a BLAST analysis , an endeavor of aligning each of millions of reads to a 3-million-nucleotide sequence. This will take some time. For the purposes of this tutorial, we shall use the Bowtie2 mapper and the Integrative Genomics Viewer (IGV) to explore the matadata for the read sequences.
For this tutorial, let us create a new session(history) and follow the steps below.
Loading Sequencing Data
An impression for loading data to a local Galaxy instance can be found in the initial part of this tutorial . You could follow the for the following data.
- https://zenodo.org/record/1324070/files/wt_H3K4me3_read1.fastq.gz
- https://zenodo.org/record/1324070/files/wt_H3K4me3_read2.fastq.gz
As noted, the data could be manually downloaded from the website and then loaded to the Galaxy instance, or the Galaxy interface provides for the direct download as well. After the files, have been loaded we shall rename these paired-end files, again, as reads_1 and reads_2 . This is the kind of data we would usually get, directly from a sequencing facility, and might be prone to standard errors of quality. Anew, the aforementined tutorial can be referred for rectifying quality issues. We won't get into the ways of mitigating quality setbacks, and join back here right after.
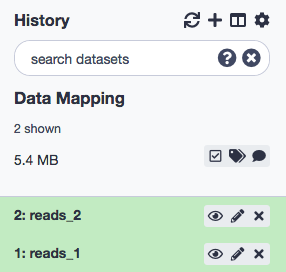
Your History Panel should probably look like this by now.
Exercise
- Run quality control measures as illustrated in the other tutorial on the paried-end reads.
Mapping data to the Reference Genome
Let us begin by reflecting on the meaning of a Reference Genome. It is a sequence of nucleotides (bases/ base-pairs) that acts as a template for a representative species. Every individual will have a unique genopme assembly, and so a reference genome is usually a chimera; henceforth it does not accord to a single organism. As the sequencing technologies advance and we further in precision and lower costs, we are tempted to sequence more individuals from a speices to enhance our biological understanding as well. This engenders us having different versions of genomes, eg. hg19, hg38 versions of the Homo sapiens, etc.
For using Bowtie2, the first step will be to install it into the Galaxy instance. Let us again go back to the tutorial enlisted here and do that first.
When the data and tool have been setup, we commence with the execution with the following paramaters.
- Paired-end: Specifying both input files
- “Do you want to set paired-end options?”: No
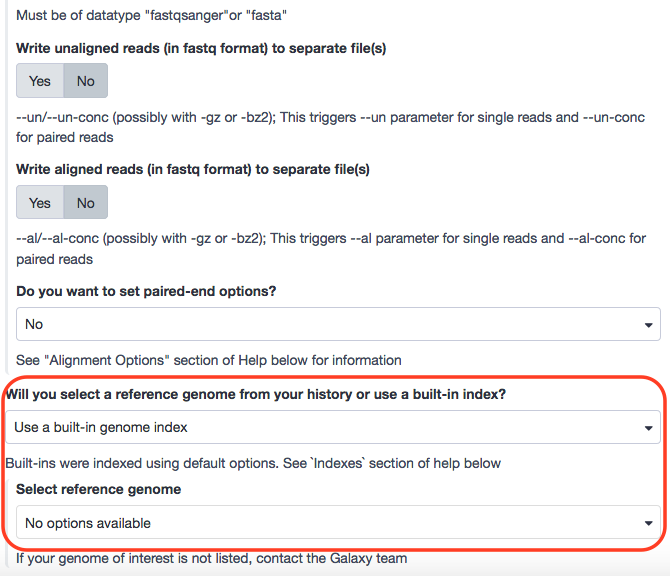
- “Will you select a reference genome from your history or use a built-in index?”: Use a built-in genome index
- “Select reference genome”: Mouse (Mus musculus): mm10
- “Select analysis mode”: Default setting only
- “Save the bowtie2 mapping statistics to the history”: Yes
Before anything though, we are confronted with a problem. The reference genome is not available in the local Galaxy.
Why so? Because the forked version of the Galaxy does not contain the "actual" data or tools, as these are completely left to the discretion of the user and so are not feasible to be accompanied with every instance of Galaxy due to requirement and memory aspects. However, these can be made available by following certain protocols.
There is an option of the rsync utility that allows a user to download data from the Main Galaxy , however, a more recommended and contemporary way to load a reference genome into a local instance of Galaxy, is to use Data Managers . These can be installed, just like any other tool, through an administrator profile. We'll focus on using data managers in this exercise.
There is also a possibility of manually uploading a genome file (.fa, .fasta), and then recalling it from session history to be used as the purported reference genome. There are two problems, albeit. First, it is not comforting to manually load reference genome and build indices for every tool that we want to use. It is just too time consuming and un-professional. We want a reference genome to be available to every tool and not just the current one. Also, an uploaded file is unique to a history. Second, the tools take a little extra time to process the external, reference genome file, as opposed to the ones that are already "build-in". Consider akin to this scenario the mobile apps that are automatically off-loaded from the system due to seldom usage. This aids memory management. Now, if you want them again, the system will download them back easily with "cached" indices. This is far efficient than installing the new ones.
A particular data manager, data_manager_fetch_genome_dbkeys_all_fasta, is required to load genome into the local instance of Galaxy. P.S. You can seek this workshop page for details. Also, bear in mind that the order of installation of the data managers is advised by the technical team of Galaxy. They recommend - retrieve fasta, index for samtools, index for picard, and then index for any other tools-in-use. Each step must be allowed to complete before proceeding.
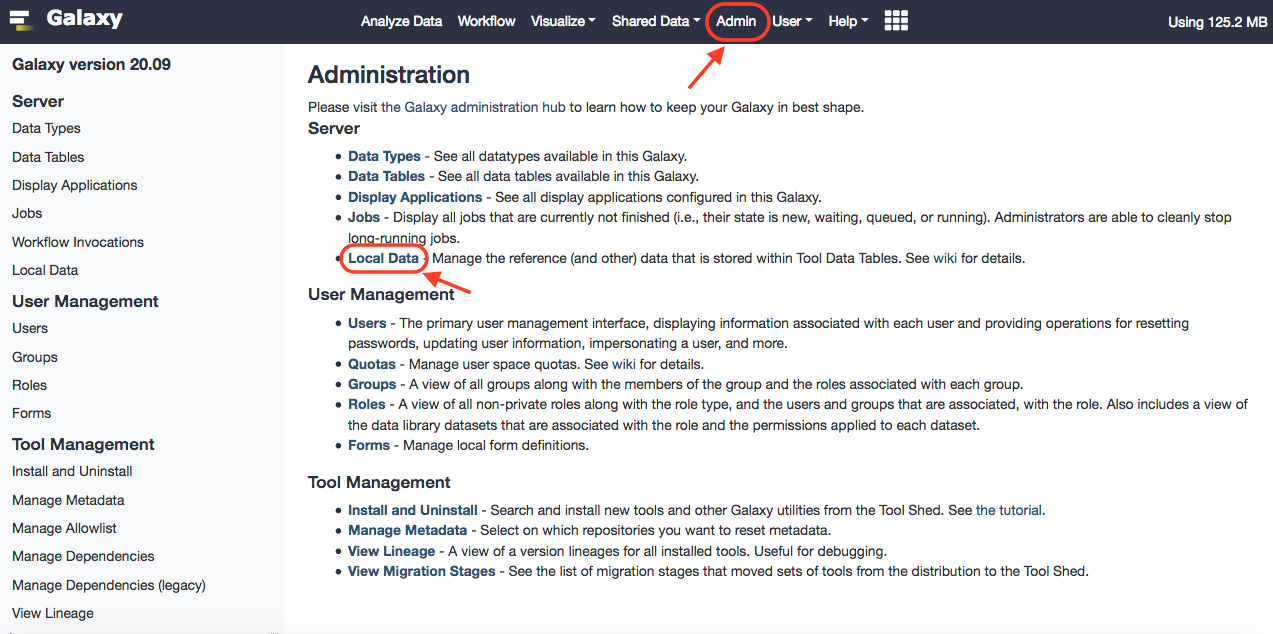
After successful installation, you can browse Local Data under Admin option of Galaxy, and reach out to the installed data manager. Upon selecting, you'll have the option to select preloaded dbkeys. These can be traced in the ~/tool-data/shared/ucsc/builds.txt file.
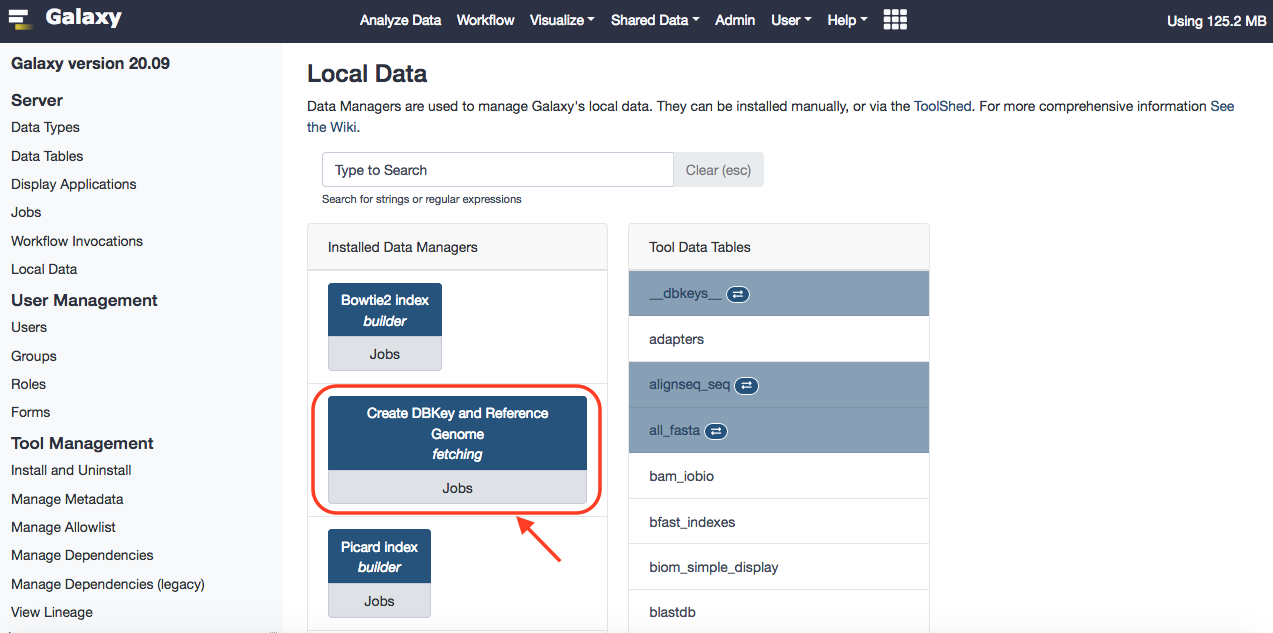
The mouse genome version "mm10" is sourced from the UCSC. Select appropriate paramaters, as under.
- "DBKEY to assign to data": Type in mm10 and select the known build from the search list results
- "Choose the source for the reference genome": UCSC
- "UCSC's DBKEY for source FASTA": mm10
- Leave the rest at default
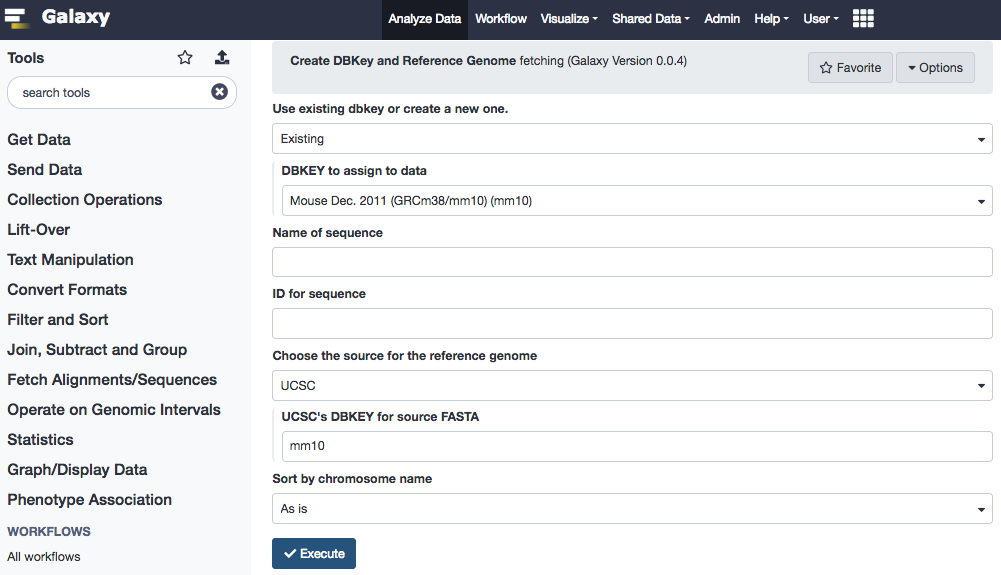
We have the reference genome now, but still we won't be able to use it. The next step is to create specific indices for the prospective tools that we wish to deploy inside the native environment of the Galaxy, so that the genome that we have installed can be referenced. Again, we shall install several other data managers, in the order as below.
- SAM indexer (Search term: data_manager_sam_fasta_index_builder )
- Picard indexer (Search term: data_manager_picard_index_builder )
- 2bit (twoBit) indexer (Search term: data_manager_twobit_builder )
These were generic indices. Now for our scenario, we would like to have the bowtie2-indexer installed. It's search term for the tool shed is data_manager_bowtie2_index_builder . Let's examine if that worked.
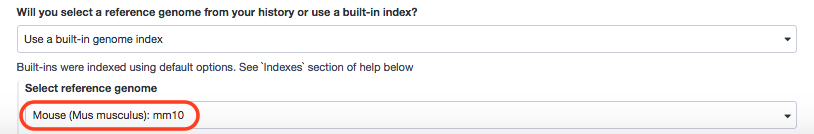
Sure enough. Next, we move to executing Bowtie2. We shall assume the following paramaters.
- “Is this single or paired library”: Paired-end
- “FASTA/Q file #1”: reads_1
- “FASTA/Q file #2”: reads_2
- “Do you want to set paired-end options?”: No
- “Will you select a reference genome from your history or use a built-in index?”: Use a built-in genome index
- “Select reference genome”: Mouse (Mus musculus): mm10
- “Select analysis mode”: Default setting only
- “Save the bowtie2 mapping statistics to the history”: Yes
You should have a look at the parameters there, specially the mate orientation if you know it. They can improve the quality of the paired-end mapping.
You should have a look at the non default parameters and try to understand them. They can have an impact on the mapping and improving it.
The tool shall output two entities- mapping stats and alignments .
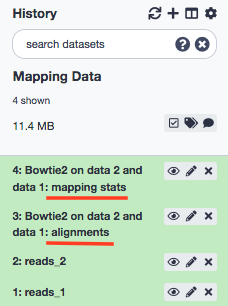
Exercise
- Contemplate the Mapping Stats from the output of Bowtie2 results.
- The result from alignment is a Binary Alignment Map (BAM) file. Discuss how is it different from a SAM file and a FASTQ (input) file.
To take the discussion further, we now install the samtools_stats repository and execute the Samtools stats function to explore the alignment results in a BAM file, i.e. output of Bowtie2.
Use the following instance.
- “BAM file”: aligned reads (output of Bowtie2 tool)
- “Use reference sequence”: Locally cached
- “Using genome”: Mouse (Mus musculus): mm10
Exercise
- Discuss the results.
For exploratory analysis, the mapped reads can be graphically visualized via browsers like, Integrative Genomics Viewer (IGV) or JBrowse .
Exercise
- Plot the output of Bowtie2 on any of these browsers and track the locations chr2:98,666,236-98,667,473 .
- Which one of the browsers do you find more easy to use?
References
- Joachim Wolff, Bérénice Batut, Helena Rasche, 2020 Mapping (Galaxy Training Materials). /training-material/topics/sequence-analysis/tutorials/mapping/tutorial.html Online; accessed Mon Jul 27 2020